Jan. 9th, 2026
Analytical labs are going through a major shift right now, moving away from traditional HPLC and into the much more sensitive world of LC-MS. This isn't just a simple detector swap. It’s a complete rethink of how we handle samples, mobile phases, and even the basic vials on the bench. For years, HPLC was the undisputed workhorse for pharmaceuticals and environmental testing, but as we started needing to find "needles in haystacks" and confirm molecular structures with 100% certainty, UV detection reached its limit. Optical detectors just can't see co-eluting peaks or hidden impurities, which is why mass spec has become the new standard. This guide walks through the technical "nitty-gritty" of this transition, focusing on why your choice of consumables is often the make-or-break factor for your data.
The Big Picture: Light Absorbance vs. Molecular Weighing
When you boil it down, the main difference between HPLC and LC-MS is how the system "sees" your sample. HPLC usually relies on UV-Vis or Fluorescence (FLD) detectors. These are essentially visual tools—they identify molecules by their chromophores, the specific parts of a structure that absorb light. While this is great for routine quality checks, it has a major blind spot: it relies solely on retention time (tR) for identification. If two different compounds elute at the exact same time, the UV detector shows one peak, and you’d never know there was an impurity hiding inside.
LC-MS changes the game by using the mass spectrometer as a "molecular scale." Instead of light, it measures the mass-to-charge ratio (m/z) of ions. By pairing chromatographic separation with mass analysis, you can distinguish between peaks even if they overlap perfectly on the column. This "dual-selectivity" is why LC-MS can pick out trace analytes in messy matrices like blood plasma or soil extracts that would be impossible to analyze with HPLC. You get the monoisotopic mass, isotopic patterns, and fragmentation data—details that no optical detector can provide.
At a Glance: How the Systems Compare
The choice between the two often comes down to how deep you need to go. HPLC is still the king of routine, high-concentration work because it's robust and cheap to run. But for metabolomics or finding tiny impurities, LC-MS is mandatory.
|
Feature
|
HPLC
|
LC-MS
|
|
Detection
|
UV-Vis Absorbance
|
Mass-to-Charge (m/z)
|
|
Primary Data
|
Retention Time, Area
|
m/z, Fragments, Area
|
|
Sensitivity
|
ug/mL down to ng/mL
|
pg/mL down to fg/mL
|
|
Selectivity
|
Moderate (Chromatographic)
|
High (Mass resolution)
|
|
Tolerance
|
High (Handles "dirty" samples)
|
Low (Prone to Ion Suppression)
|
|
Buffers
|
Flexible (Non-volatile okay)
|
Strict (Must be Volatile)
|
|
Best Use Case
|
Raw materials, Purity QC
|
Proteomics, Trace analysis
|
The Sodium Adduct Headache: Why Cheap Vials Kill Your Data
One of the biggest hurdles when moving to LC-MS is the chemical "noise" coming from your consumables, especially your glassware. In the HPLC world, we rarely think about the vial unless we’re dealing with a very basic sample. But in LC-MS, trace amounts of sodium (Na+) leaching out of the glass can completely change your mass spectrum, making it look like your molecule is something else entirely.
The [M+Na]+ Interference Problem
Most LC-MS systems use Electrospray Ionization (ESI), which turns liquid molecules into gas-phase ions. Usually, we want to see protonated molecules ([M+H]+). However, sodium loves oxygen-rich groups like carboxyls and carbonyls. When sodium ions are present—even in tiny amounts—they compete with protons to form sodium adducts. These show up as [M+Na]+ peaks at a mass shift of +22.99 Da. To eliminate this leaching, using high-quality
9mm short thread vials with a wide opening
is a critical step in maintaining baseline purity.
.png)
This causes three major problems:
-
Signal Splitting: Your analyte signal gets divided between [M+H]+, [M+Na]+, and maybe even [M+K]+, which tanks your sensitivity.
-
Fragmentation Failures: Sodium adducts are way more stable than protonated ones. They don't break apart easily in MS/MS, which means they are useless for MRM or quantitative work.
-
Ghost Peaks: In untargeted work, sodium clusters can fill your baseline with "ghost peaks" that mask the real biomarkers you're looking for.
Why Glass Leaches (The pH Effect)
The sodium usually comes from standard borosilicate glass. Glass isn't as inert as we like to think; it goes through a pH-dependent ion exchange with your sample. Hydronium ions (H3O+) from your diluent move into the glass and push sodium ions (Na+) out into your sample:
Si-O-Na + H3O+ --> Si-OH + Na+ + H2O
This makes your sample more alkaline—sometimes the pH will spike above 9.0. When that happens, hydroxyl ions (OH-) start attacking the silica network (the Si-O-Si bonds) itself, dissolving the glass and dumping even more sodium and silicate into your vial. This is why LC-MS grade vials are a must. They are made from high-purity Type I borosilicate glass and are treated to ensure almost zero leaching, keeping your pH neutral and your baseline clean.
Dealing with Bubbles: Stable Ions Start with Clean Degassing
In HPLC, a bubble usually just causes a baseline spike or a pump error. In LC-MS, it’s a disaster. Even a microscopic bubble can destabilize the ESI source, causing your signal to flicker or drop out entirely.
The Physics of the Taylor Cone
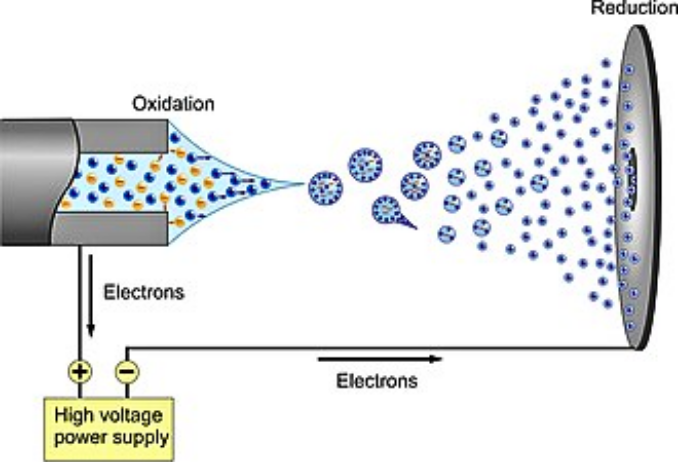
ESI stability depends on a stable Taylor cone at the needle tip. This cone is a liquid meniscus held in place by a delicate balance of surface tension and electric fields. From the tip of this cone, you get a steady spray of charged droplets. If a gas bubble reaches that needle, it changes the surface tension instantly. The Taylor cone starts to pulsate or collapses, and your ion current goes haywire.
Studies with high-speed cameras show that bubbles disrupt the "cone-jet" mode and force the spray into an erratic, non-axial pattern. This means the droplets come off too large to evaporate properly, and your sensitivity drops off a cliff.
Why Sonication is "Disappointing"
A lot of old-school HPLC methods tell you to sonicate your mobile phase. But for LC-MS, sonication is remarkably poor. It only removes about 20% to 30% of dissolved gases. Even worse, as soon as you stop sonicating, the solvent starts soaking up air again from the room.
Modern LC-MS labs use in-line vacuum degassers. These run the mobile phase through a gas-permeable membrane under a vacuum right before it hits the pump. They are much more efficient, removing up to 99% of dissolved gases and keeping your solvents bubble-free throughout the entire run. For trace work where you need a rock-solid signal, in-line degassing is mandatory,using
universal laboratory syringe filters
during sample preparation is the industry standard.
Peak Integrity: Dwell Volume and Extra-Column Broadening
When you move a method from HPLC to LC-MS, you might see your peaks get "fatter" or lose resolution. This is usually due to dwell volume and extra-column volume—things we often ignore in HPLC but that dominate LC-MS because we use smaller columns and lower flows.
Dwell Volume (The Gradient Delay)
Dwell volume is the total space between where your solvents mix and where they hit the column. This volume creates a delay. If you’re running a 2.1 mm column at 200 uL/min and you have a 400 uL dwell volume, your column won’t even "see" the gradient for the first 2 minutes of the run. This shift in timing can change how your early peaks elute and can even cause them to overlap.
Extra-Column Volume (The Mixing Effect)
While dwell volume affects timing, extra-column volume (everything from the injector to the detector) affects the peak width. In LC-MS, the tubing connecting the column to the MS source is the biggest culprit for peak broadening.
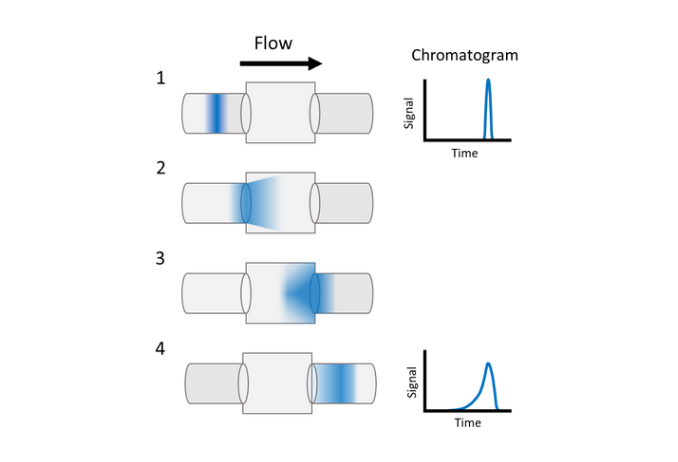
This happens because of dispersion—the liquid in the center of the tube moves faster than the liquid at the walls. This is especially bad in LC-MS because we use narrow-bore tubing and lower flow rates to help ESI efficiency. Also, if you use a UV detector and an MS in series, the UV flow cell and the extra tubing act like a "mixer," blurring your peaks before they ever reach the mass spec.
To keep your peaks sharp, use the shortest possible lengths of 0.1 mm or 0.127 mm ID tubing and high-precision fittings (like Agilent "Viper" styles) to eliminate any "dead space" where samples can hang out.
The LC-MS Upgrade Checklist: What to Swap
If you're making the jump to LC-MS, you need to systematically upgrade your materials.
-
Solvents: Toss the "HPLC Grade" and get "LC-MS Grade." These are certified for ultra-low trace metals (Na+, K+) so you don't get adducts.
-
Buffers: Stop using Phosphates immediately. Only use volatile buffers like Formic Acid, Acetic Acid, or Ammonium Acetate.
-
Vials: Use Certified Low-Background (LC-MS Grade)
9mm short thread vials
to stop sodium leaching.
-
Caps & Septa: Ensure a perfect seal with
9mm short thread screw caps with bonded septa
to prevent contamination from adhesives.
-
Filtration: Switch from 0.45 um to 0.22 um filters. Use
syringe filters and membranes
optimized for low extractables.
-
Tubing: Use 0.1 mm or 0.127 mm ID tubing and zero-dead-volume (ZDV) fittings to keep your chromatography tight.

LC-MS vs. GC-MS: A Quick Comparison
|
Factor
|
GC-MS
|
LC-MS
|
|
Analyte
|
Volatile compounds
|
Non-volatile, polar, or big molecules
|
|
Stability
|
Must handle high heat
|
Works at room temperature
|
|
Mobile Phase
|
Carrier gas (Helium/Nitrogen)
|
Solvents (ACN/Water/MeOH)
|
|
Sample Prep
|
Often needs derivatization
|
Usually simple dilution/filtration
|
Lab Terminology Cheat Sheet (Quick Reference)
|
English
|
Persian (فارسی)
|
Russian (Русский)
|
|
Analyte
|
آنالیت
|
Аналит
|
|
Mobile Phase
|
فاز متحرک
|
Подвижная фаза
|
|
Stationary Phase
|
فاز ساکن
|
Неподвижная фаза
|
|
Retention Time
|
زمان بازداری
|
Время удерживания
|
|
Sensitivity
|
حساسیت
|
Чувствительность
|
|
Specificity
|
اختصاصی بودن
|
Специфичность
|
|
Dwell Volume
|
حجم تاخیر
|
Объем задержки
|
|
Peak Broadening
|
پهن شدن پیک
|
Уширение пика
|
|
Adduct
|
ادکت
|
Аддукт
|
|
Degassing
|
گاززدایی
|
Дегазация
|
FAQ: Real-World Questions from the Lab
1. Why do I see [M+23]+ peaks in LC-MS but not in HPLC? HPLC only sees light absorbance, and sodium doesn't absorb UV. But LC-MS sees everything that ionizes. Sodium leached from glass or present in poor-quality water forms a [M+Na]+ adduct, which is the mass of your molecule plus 22.99 Da.
2. Can I use phosphate buffers if I filter them really well? No. Filters only remove particles. Phosphate is a non-volatile salt. In the ESI source, the liquid evaporates, but the salt stays behind as solid crystals. These will clog your ESI needle and the sampling orifice, eventually killing your instrument.
3. What's the real difference between HPLC and LC-MS grade water? LC-MS grade water is tested specifically for trace metals like sodium and potassium. Using HPLC water in an LC-MS often results in high background noise and constant sodium adduct problems.
4. Why is my retention time different on my new LC-MS system? It's likely the dwell volume. If your new system has a different volume between the mixer and the column than your old HPLC, the gradient will hit the column at a different time, shifting your peaks.
5. Is sonication enough to degas my LC-MS solvents? Not really. Sonication only gets rid of about 20-30% of dissolved gases. In-line vacuum degassers are much better (up to 99%) and continuous, which is what you need to keep the ESI Taylor cone stable.
Conclusion: Mastering the Transition
Moving from HPLC to LC-MS is a massive upgrade in power, but it requires a much higher standard for purity and precision. The "HPLC vs. LC-MS" difference isn't just about the detector; it’s about the physics of the Taylor cone and the subtle chemistry of your vials. By focusing on chemical purity (LC-MS vials and solvents), fluidic optimization (minimal tubing volume), and physical stability (in-line degassing), you can ensure your transition is a success and your data is as sharp and accurate as possible.
 English
English
 Chinese
Chinese
